
A medical device is an item that treats, cures, or prevents certain diseases and health conditions. Doctors and medical professionals use them in hospitals and outpatient facilities, and consumers can buy devices to use at home. Since they are so crucial for good health, manufacturers must follow strict medical device labeling requirements, including FDA medical device labeling requirements and other global medical device labeling standards.
Medical devices have specific purposes, so the label must adequately share all relevant information. A label should show how to use the device and also give transparent details about the manufacturer. Clear labeling supports safety, regulatory compliance, and consistent quality across medical device packaging requirements.
Types of Labels Required
The Food and Drug Administration (FDA) helps manufacturers classify medical devices by determining their purpose or searching a database for similar items. From there, you’ll understand what labels you need to put on your device.
As requirements vary depending on the device, checking the regulations before selling products is crucial for compliance. Industries that outsource medical device assembly should verify labels follow FDA regulations before putting their products on the market.
Product Labeling
Product labeling must present key information required by regulators and by medical device labeling standards. These standards include FDA labeling requirements for medical devices, ISO 13485, and additional labeling regulatory requirements for medical devices that help ensure product safety and traceability.
FDA regulations require that manufacturers print labels on products that include:
-
Brand name
-
Device identifier
-
Details about the production lot
-
Expiration date
-
Storage and handling instructions
-
Unique device identifier (UDI)
The information on the product ensures that consumers can get help even if they lose the external packaging and instructions. There is enough information about the manufacturer and support that they can use the UDI to access the lost information.
Manufacturers link the UDI to the GUDID database, which the FDA oversees. This process plays a major role in labeling regulatory requirements for medical devices and ensures consistent traceability across medical device label examples used by healthcare organizations.
Packaging Labeling
Package labeling must include usage instructions, storage guidance, and manufacturer details. These details directly support medical device packaging standards and medical device packaging requirements that help ensure the product is used safely.
External packaging should also clearly display UDI. This links the device to its packaging and supports compliance with medical device packaging regulations and long-term traceability.
Information Included on Labels
Medical device manufacturers need to include critical information on both the device and its packaging to maintain compliance with FDA medical device labeling requirements and medical device labeling regulatory requirements. Typical label content includes:
-
Product name
-
Manufacturer name and address
-
Customer support phone number or website
-
Model/catalog or part number
-
Serial number (if applicable)
-
UDI (human-readable and barcode)
-
CE marking, if relevant
These elements help ensure device accuracy, prevent counterfeit risks, and support global medical device labeling requirements for safety and recalls.
Additional Labeling Requirements Depending on Device Type
Regulatory demands grow more specific depending on the class of the device:
-
Class I Devices: Lower-risk devices require basic labeling but must still meet essential medical device labeling requirements.
-
Class IIa / IIb and Class III Devices: As the device class increases, FDA labeling requirements for medical devices and ISO 13485 labeling standards become more detailed and demanding.
-
In Vitro Diagnostic (IVD) Devices: Specialized labeling is required, including UDI and clear usage instructions. These follow medical device labeling requirements and medical device packaging standards for traceability and safe handling.
Best Practices for Maintaining Labeling Compliance
To consistently meet medical device labeling standards and medical device packaging regulations, manufacturers should:
1. Risk Management Plan
Develop a risk management plan that considers how labeling impacts patient safety and device handling. This supports both FDA medical device labeling requirements and ISO labeling expectations.
2. Quality Management System (QMS)
A strong QMS ensures labeling is accurate and consistent. Using an ISO 13485 QMS helps maintain compliance with medical device labeling requirements and medical device packaging requirements over the full product lifecycle.
Why These Labeling Practices Matter
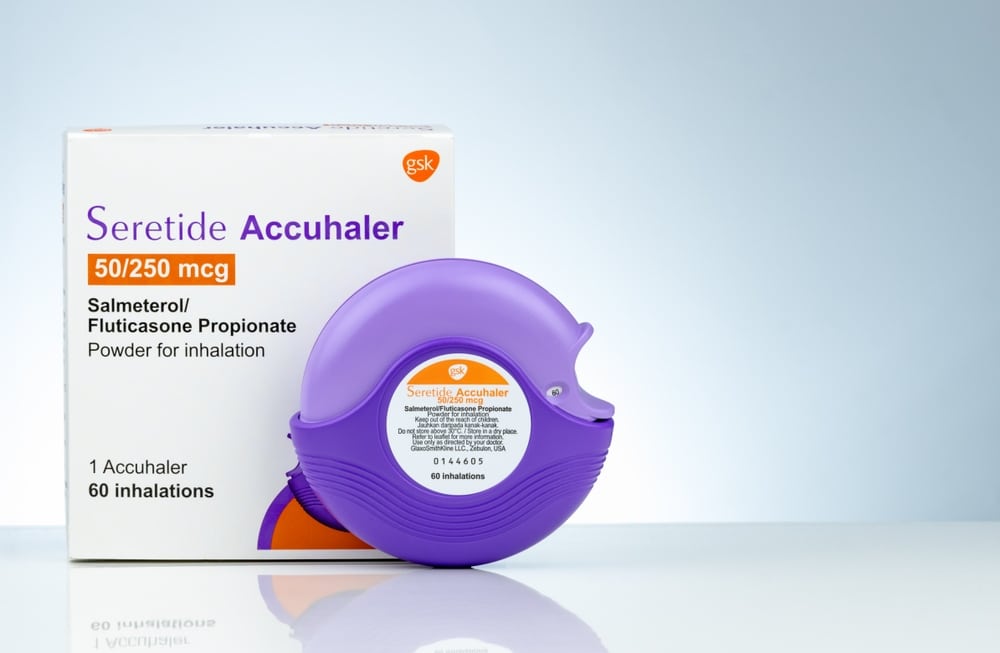
Accurate labeling helps ensure safe use, prevents errors, and supports global compliance across medical device packaging regulations and FDA medical device labeling requirements. This not only protects users but also strengthens trust with healthcare partners.
Clear labeling also reduces rework and supports smoother inspections, helping manufacturers meet deadlines so patients can get the care they need.
Learn More About Medical Device Assembly & Packaging
If you need help meeting medical device labeling requirements or aligning with medical device packaging standards, MDI provides structured, ISO-certified support. Our white room environment helps maintain quality so your products are delivered safely. The only thing safer than your device in our workspace is your reputation in our hands.
Read more about MDI’s Medical Device Assembly & Packaging Service →
https://mdi.org/services/medical-device-assembly-services/